关键信息
-
基因名
RBBP4
- 应用
-
别名
RBBP4;RBAP48;Histone-binding Protein RBBP4
-
种属
Human
-
表达系统
E. coli
-
标签
His tag N-Terminus
-
纯度
Greater than 90% as determined by SDS-PAGE.
-
蛋白编号
Q09028
-
表达区间
2-425aa
-
氨基酸序列
ADKEAAFDD AVEERVINEE YKIWKKNTPF LYDLVMTHAL EWPSLTAQWL PDVTRPEGKD FSIHRLVLGT HTSDEQNHLV IASVQLPNDD AQFDASHYDS EKGEFGGFGS VSGKIEIEIK INHEGEVNRA RYMPQNPCII ATKTPSSDVL VFDYTKHPSK PDPSGECNPD LRLRGHQKEG YGLSWNPNLS GHLLSASDDH TICLWDISAV PKEGKVVDAK TIFTGHTAVV EDVSWHLLHE SLFGSVADDQ KLMIWDTRSN NTSKPSHSVD AHTAEVNCLS FNPYSEFILA TGSADKTVAL WDLRNLKLKL HSFESHKDEI FQVQWSPHNE TILASSGTDR RLNVWDLSKI GEEQSPEDAE DGPPELLFIH GGHTAKISDF SWNPNEPWVI CSVSEDNIMQ VWQMAENIYN DEDPEGSVDP EGQGS
-
内毒素
< 1.0 EU per μg protein as determined by the LAL method.
-
性状
Freeze-dried powder
-
缓冲液
PBS, pH7.4, containing 0.01% SKL, 1mM DTT, 5% Trehalose and Proclin300.
-
复溶方法
Reconstitute in ddH2O to a concentration of 0.1-0.5 mg/mL. Do not vortex.
- 个性化定制
-
稳定性测试
The thermal stability is described by the loss rate. The loss rate was determined by accelerated thermal degradation test, that is, incubate the protein at 37℃ for 48h, and no obvious degradation and precipitation were observed. The loss rate isless than 8% within the expiration date under appropriate storage condition.
-
保存条件 & 期限
Samples are stable for up to twelve months from date of receipt at -20℃ to -80℃. Store it under sterile conditions at -20℃ to -80℃. It is recommended that the protein be aliquoted for optimal storage. Avoid repeated freeze-thaw cycles.
-
运输条件
In general, recombinant proteins are supplied as lyophilized powder and shipped at ambient temperature. For bulk packages, the proteins are provided as frozen liquid and shipped with blue ice, unless otherwise requested by the customer.
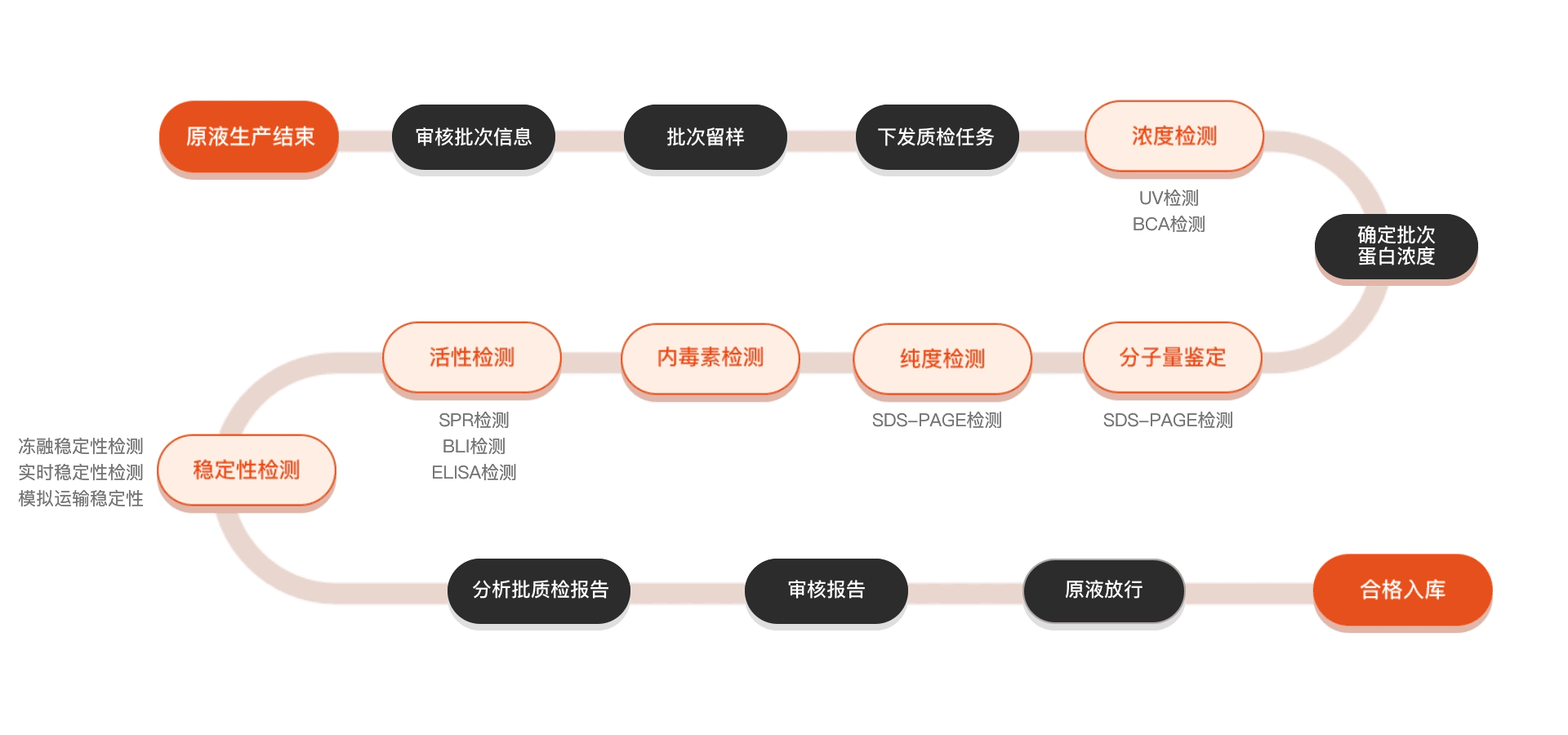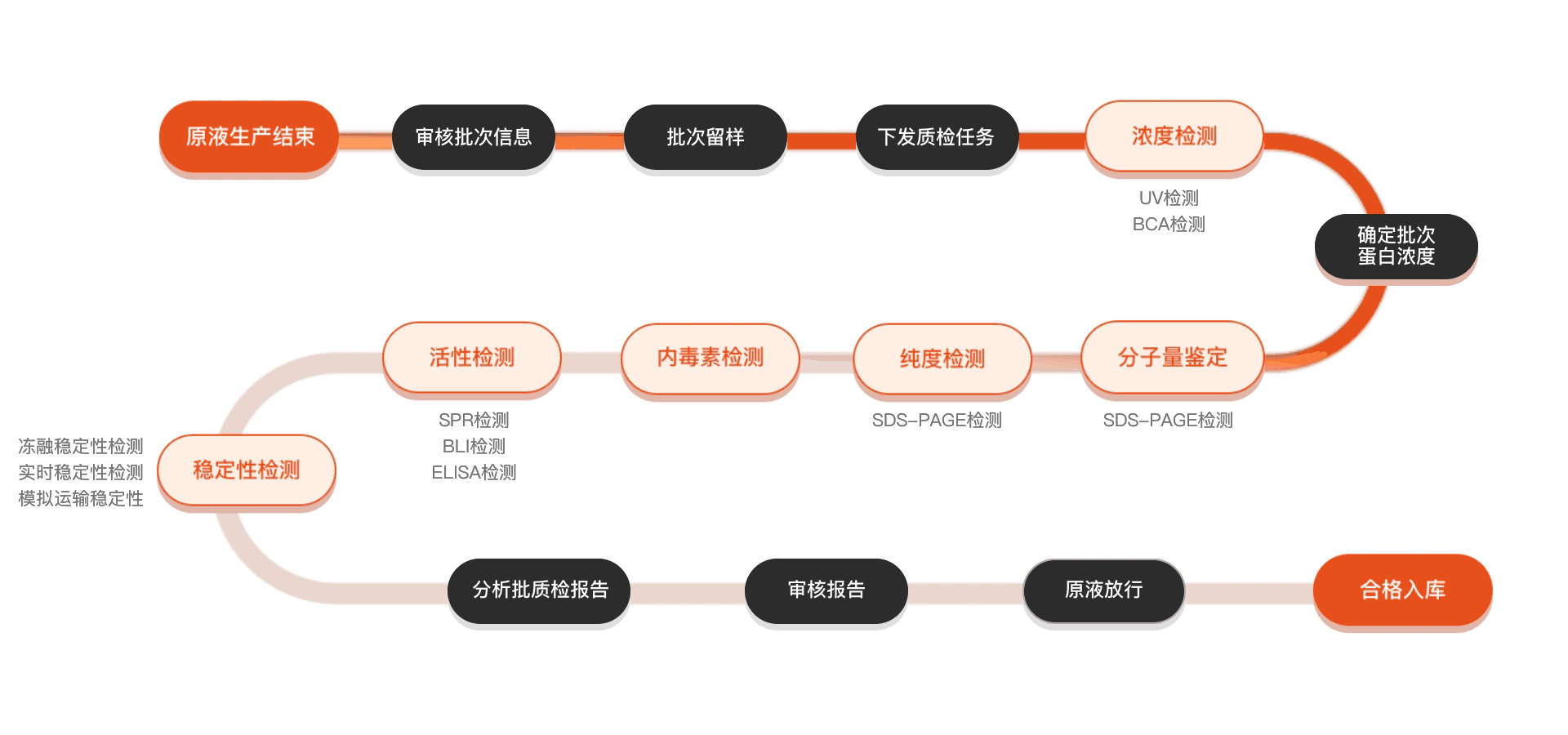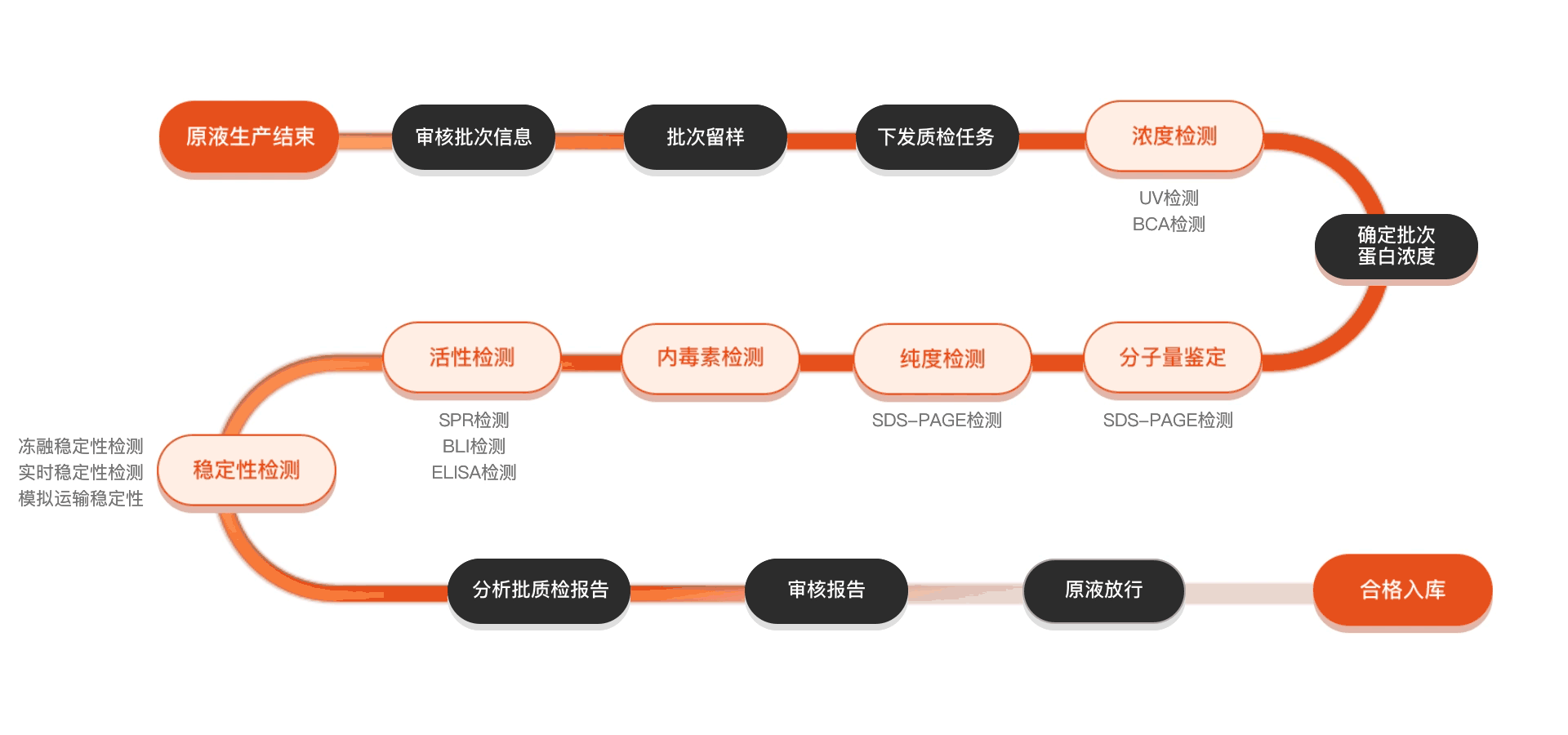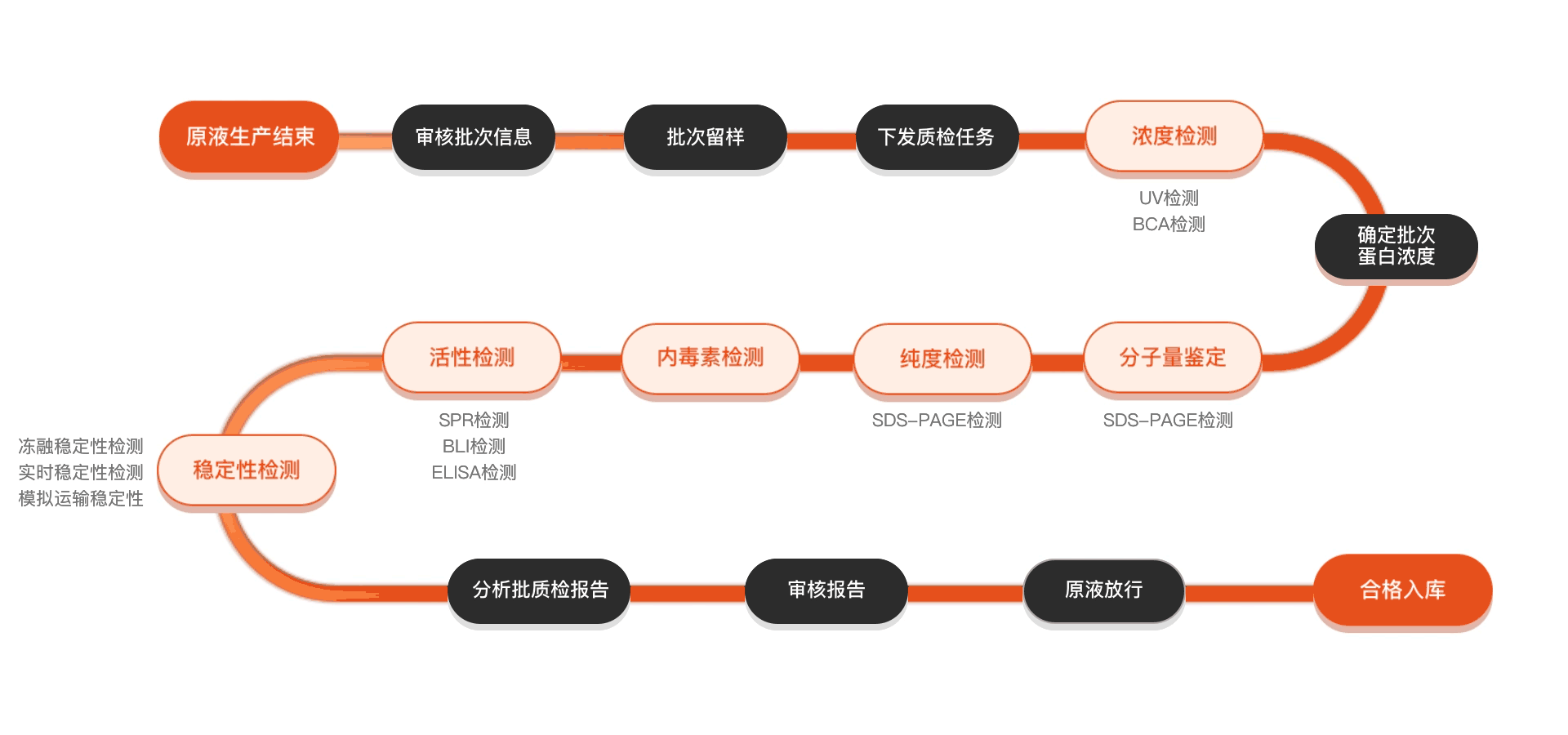
质检流程
相关产品






背景信息
RBBP4, or Retinoblastoma Binding Protein 4, is a crucial component of the chromatin remodeling machinery and plays a significant role in regulating gene expression, cell cycle progression, and differentiation. Its involvement in the retinoblastoma tumor suppressor pathway highlights its importance in cancer research, particularly in understanding mechanisms of tumorigenesis. Recent studies have demonstrated that RBBP4 interacts with various transcription factors and chromatin remodeling complexes, suggesting its multifaceted role in cellular processes. Additionally, aberrant expression or mutations of RBBP4 have been linked to several types of cancers, implying its potential as a biomarker for cancer diagnosis and prognosis. The recombinant expression of RBBP4 allows for detailed studies of its biochemical properties, functional interactions, and role in chromatin dynamics. These investigations can provide insights into its mechanisms of action, paving the way for the development of novel therapeutic strategies targeting RBBP4 in cancer therapy and other diseases where dysregulation of chromatin structure is implicated. Overall, RBBP4 research continues to be a promising avenue in understanding fundamental biological processes and potential applications in medical science.