关键信息
-
基因名
IL-12 beta
-
简介
IL-23 and IL12B together constitute the pro-inflammatory cytokine IL-23, which is crucial in innate and adaptive immunity. IL-23 is released by antigen-presenting cells such as dendritic cells or macrophages, binds to IL12RB1 and IL23R, and activates JAK2 and TYK2. Human IL-23 alpha & Mouse IL-12 beta Heterodimer Protein (HEK293, His) is a recombinant protein dimer complex containing mouse, human-derived Human IL-23 alpha & Mouse IL-12 beta Heterodimer protein, expressed by HEK293 , with C-6*His labeled tag.
- 应用
-
生物活性
1. Measured by its ability to induce IL-17 secretion by CTLL-2 cells. The ED50 for this effect is 2.703 ng/mL, corresponding to a specific activity is 3.7×10^5 U/mg. 2. Loaded Picankibart (HY-P990057) on AHC2 biosensor, can bind Human IL-23 alpha & Mouse IL-12 beta Heterodimer Protein (HEK293, His) with an affinity constant of 7.207E-11 M as determined in BLI assay. Measured by its ability to induce IL-17 secretion by CTLL-2 cells.The ED50 for this effect is 2.703 ng/mL, corresponding to a specific activity is 3.7×105 U/mg. Loaded Picankibart (HY-P990057) on AHC2 biosensor, can bind Human IL-23 alpha & Mouse IL-12 beta Heterodimer Protein (HEK293, His) with an affinity constant of 7.207E-11 M as determined in BLI assay.
-
别名
IL-12; Interleukin 12; Interleukin-12 subunit alpha; IL-12A; Cytotoxic lymphocyte maturation factor 35 kDa subunit; CLMF p35; IL-12 subunit p35; Interleukin-12 subunit beta; IL-12B; Cytotoxic lymphocyte maturation factor 40 kDa subunit; CLMF p40; IL-12 subunit p40
-
种属
Human;Mouse
-
表达系统
HEK293
-
标签
C-6*His
-
纯度
Greater than 95% as determined by reducing SDS-PAGE. as determined by SDS-PAGE.
-
蛋白编号
Q9NPF7 (R20-P189)&P43432
-
表达区间
Q9NPF7 (R20-P189)&P43432 (M23-S335)
-
氨基酸序列
RAVPGGSSPAWTQCQQLSQKLCTLAWSAHPLVGHMDLREEGDEETTNDVPHIQCGDGCDPQGLRDNSQFCLQRIHQGLIFYEKLLGSDIFTGEPSLLPDSPVGQLHASLLGLSQLLQPEGHHWETQQIPSLSPSQPWQRLLLRFKILRSLQAFVAVAARVFAHGAATLSP&MWELEKDVYVVEVDWTPDAPGETVNLTCDTPEEDDITWTSDQRHGVIGSGKTLTITVKEFLDAGQYTCHKGGETLSHSHLLLHKKENGIWSTEILKNFKNKTFLKCEAPNYSGRFTCSWLVQRNMDLKFNIKSSSSSPDSRAVTCGMASLSAEKVTLDQRDYEKYSVSCQEDVTCPTAEETLPIELALEARQQNKYENYSTSFFIRDIIKPDPPKNLQMKPLKNSQVEVSWEYPDSWSTPHSYFSLKFFVRIQRKKEKMKETEEGCNQKGAFLVEKTSTEVQCKGGNVCVQAQDRYYNSSCSKWACVPCRVRS
-
分子量
23.18 & 40-50 kDa
-
内毒素
< 1.0 EU per μg protein as determined by the LAL method.
-
性状
Freeze-dried powder
-
缓冲液
PBS, pH7.4, containing 0.01% SKL, 1mM DTT, 5% Trehalose and Proclin300.
-
复溶方法
Reconstitute in ddH2O to a concentration of 0.1-0.5 mg/mL. Do not vortex.
- 个性化定制
-
稳定性测试
The thermal stability is described by the loss rate. The loss rate was determined by accelerated thermal degradation test, that is, incubate the protein at 37℃ for 48h, and no obvious degradation and precipitation were observed. The loss rate isless than 8% within the expiration date under appropriate storage condition.
-
保存条件 & 期限
Samples are stable for up to twelve months from date of receipt at -20℃ to -80℃. Store it under sterile conditions at -20℃ to -80℃. It is recommended that the protein be aliquoted for optimal storage. Avoid repeated freeze-thaw cycles.
-
运输条件
In general, recombinant proteins are supplied as lyophilized powder and shipped at ambient temperature. For bulk packages, the proteins are provided as frozen liquid and shipped with blue ice, unless otherwise requested by the customer.
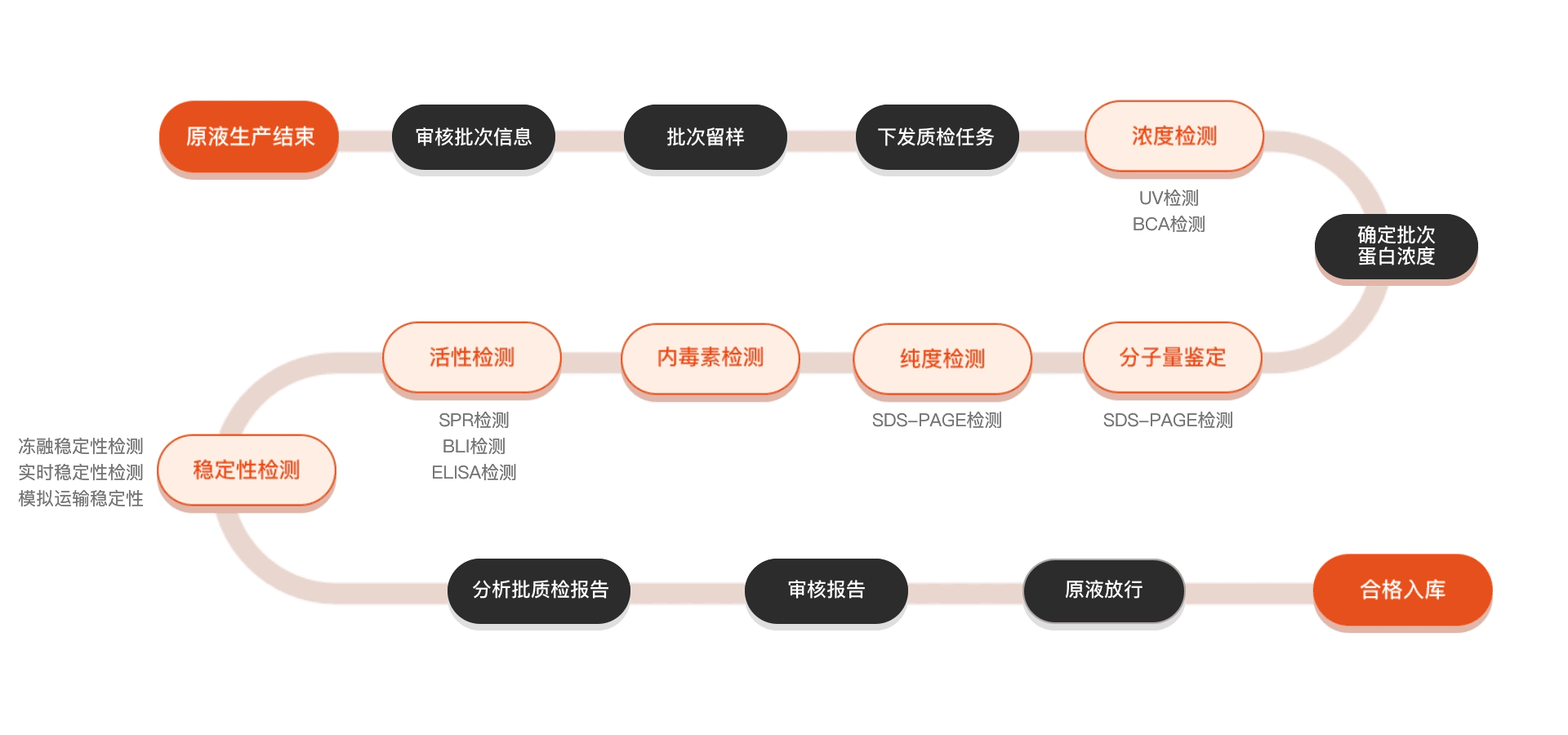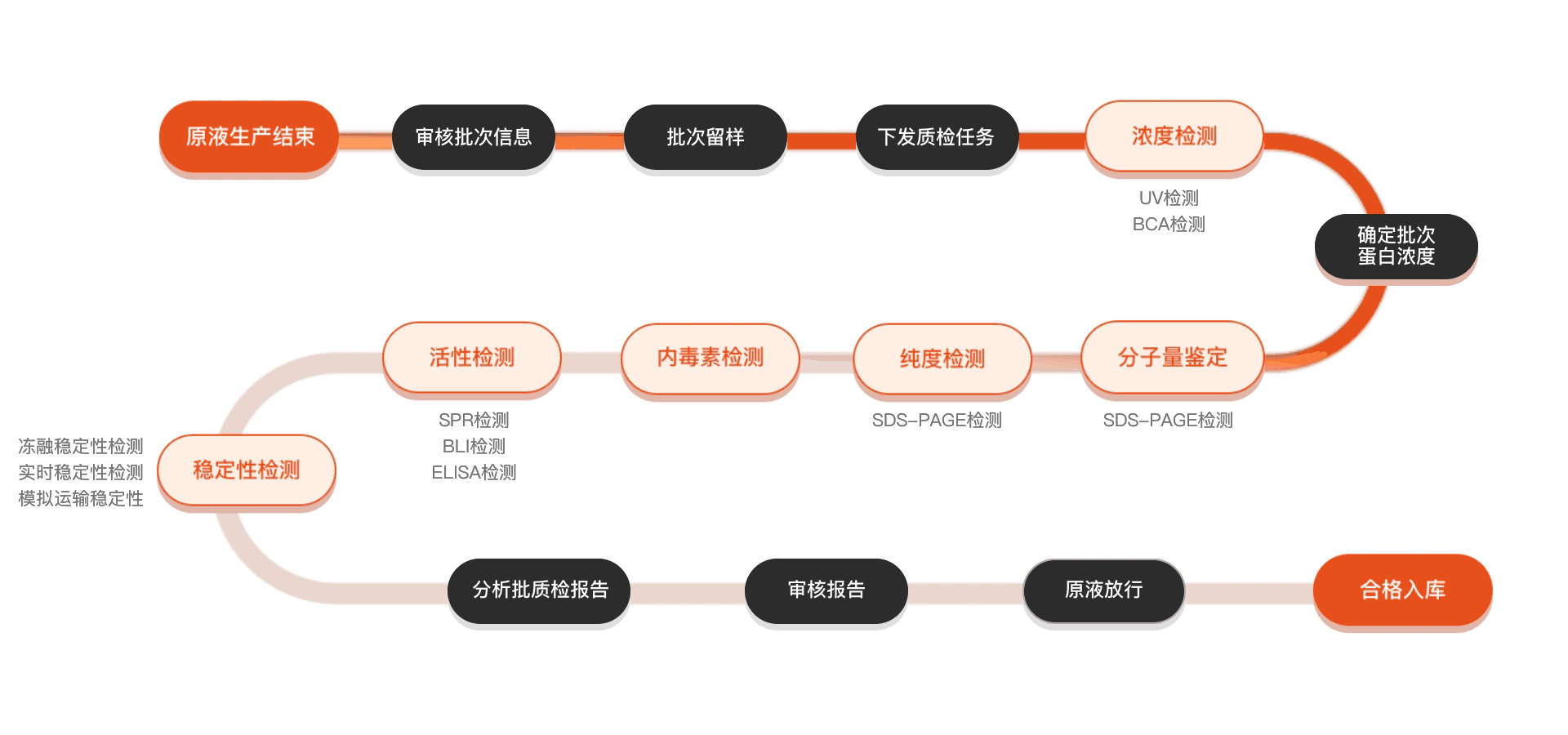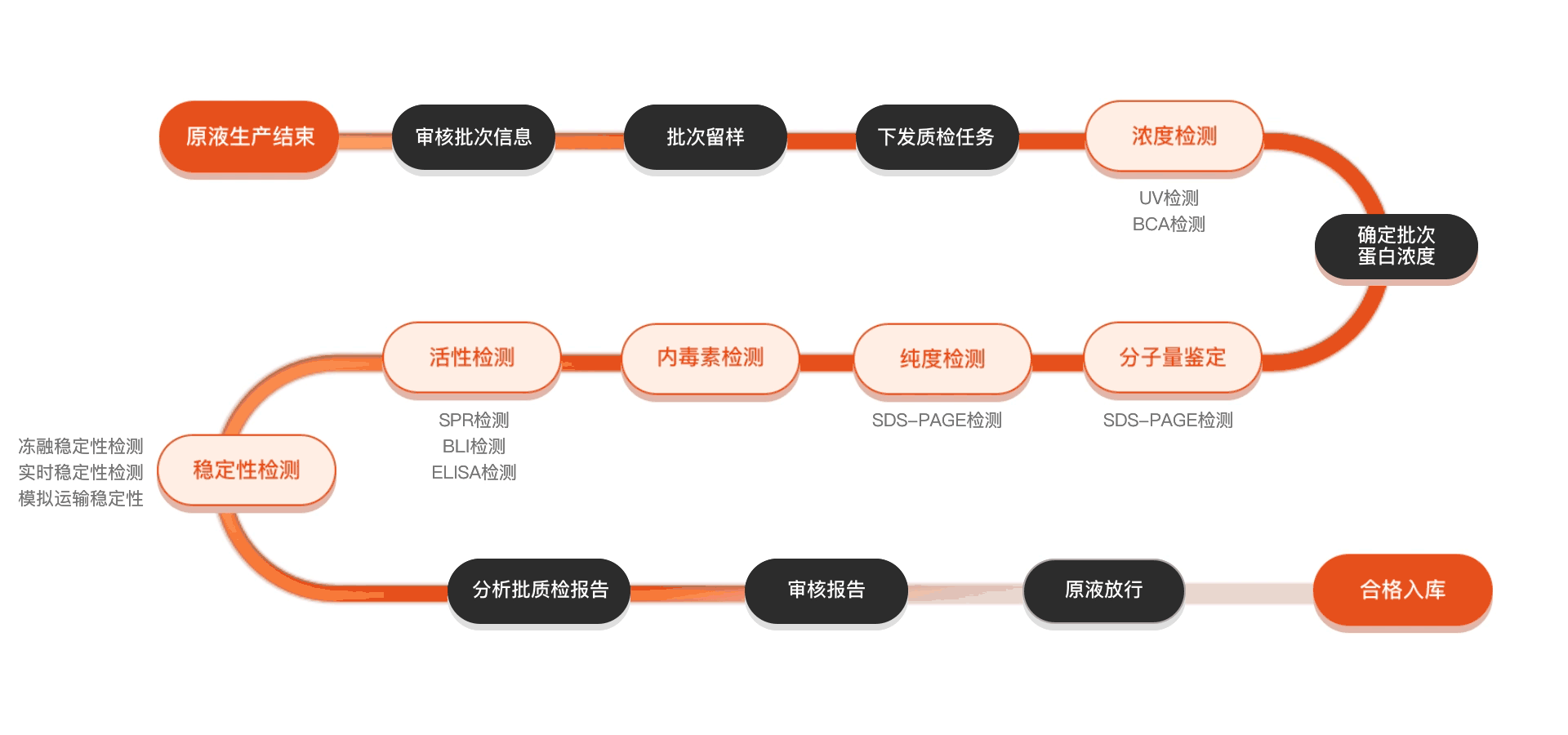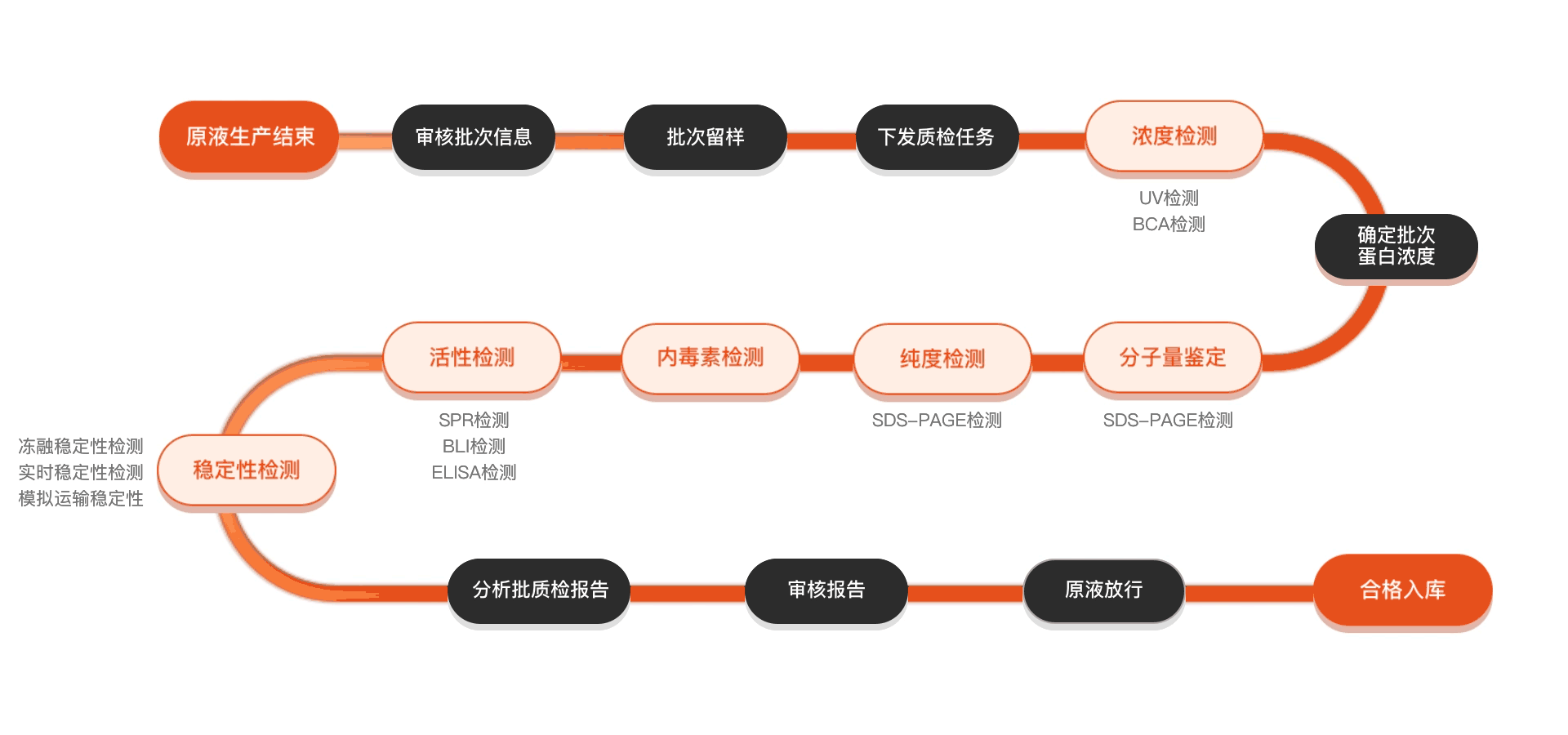
质检流程
相关产品






背景信息
Interleukin-12 (IL-12) is a pivotal cytokine that plays a crucial role in immune regulation and host defense against pathogens and tumors. IL-12 is a heterodimeric protein composed of two subunits, p35 and p40, encoded by separate genes. The p40 subunit, often referred to as IL-12 beta, has garnered significant research interest due to its involvement in immune responses. IL-12 promotes T cell differentiation into Th1 cells, enhances the production of interferon-gamma, and stimulates cytotoxic T lymphocytes and natural killer (NK) cell activity, making it essential for combating intracellular pathogens and malignancies. Dysfunctional IL-12 signaling is implicated in various autoimmune diseases and cancers, further highlighting its relevance in immunotherapy applications. Recombinant IL-12 beta protein has been developed for therapeutic purposes, including cancer vaccines and immune modulation strategies. Studies have demonstrated that delivering IL-12 beta can enhance anti-tumor immunity and improve treatment outcomes in preclinical and clinical settings. Consequently, ongoing research is focused on optimizing the production, delivery mechanisms, and therapeutic efficacy of recombinant IL-12 beta, aiming to harness its potential to improve cancer therapies and address unmet medical needs in immunological disorders.