关键信息
-
基因名
RPL13
- 应用
-
别名
60S ribosomal protein L13; BBC1; Breast basic conserved gene 1; Breast basic conserved protein 1; D16S444E ; D16S44E; FLJ27453 ; FLJ27454 ; L13; MGC117342 ; MGC71373 ; OK/SW cl.46; OK/SWcl.46; Ribosomal protein L13; RL13_HUMAN; rpl13
-
种属
Human
-
表达系统
E. coli
-
标签
His tag N-Terminus
-
纯度
Greater than 90% as determined by SDS-PAGE.
-
蛋白编号
P26373
-
表达区间
1-211 aa
-
氨基酸序列
MAPSRNGMVL KPHFHKDWQR RVATWFNQPA RKIRRRKARQ AKARRIAPRP ASGPIRPIVR CPTVRYHTKV RAGRGFSLEE LRVAGIHKKV ARTIGISVDP RRRNKSTESL QANVQRLKEY RSKLILFPRK PSAPKKGDSS AEELKLATQL TGPVMPVRNV YKKEKARVIT EEEKNFKAFA SLRMARANAR LFGIRAKRAK EAAEQDVEKK K
-
分子量
24.2 kDa
-
内毒素
< 1.0 EU per μg protein as determined by the LAL method.
-
性状
Freeze-dried powder
-
缓冲液
PBS, pH7.4, containing 0.01% SKL, 1mM DTT, 5% Trehalose and Proclin300.
-
复溶方法
Reconstitute in ddH2O to a concentration of 0.1-0.5 mg/mL. Do not vortex.
- 个性化定制
-
稳定性测试
The thermal stability is described by the loss rate. The loss rate was determined by accelerated thermal degradation test, that is, incubate the protein at 37℃ for 48h, and no obvious degradation and precipitation were observed. The loss rate isless than 8% within the expiration date under appropriate storage condition.
-
保存条件 & 期限
Samples are stable for up to twelve months from date of receipt at -20℃ to -80℃. Store it under sterile conditions at -20℃ to -80℃. It is recommended that the protein be aliquoted for optimal storage. Avoid repeated freeze-thaw cycles.
-
运输条件
In general, recombinant proteins are supplied as lyophilized powder and shipped at ambient temperature. For bulk packages, the proteins are provided as frozen liquid and shipped with blue ice, unless otherwise requested by the customer.
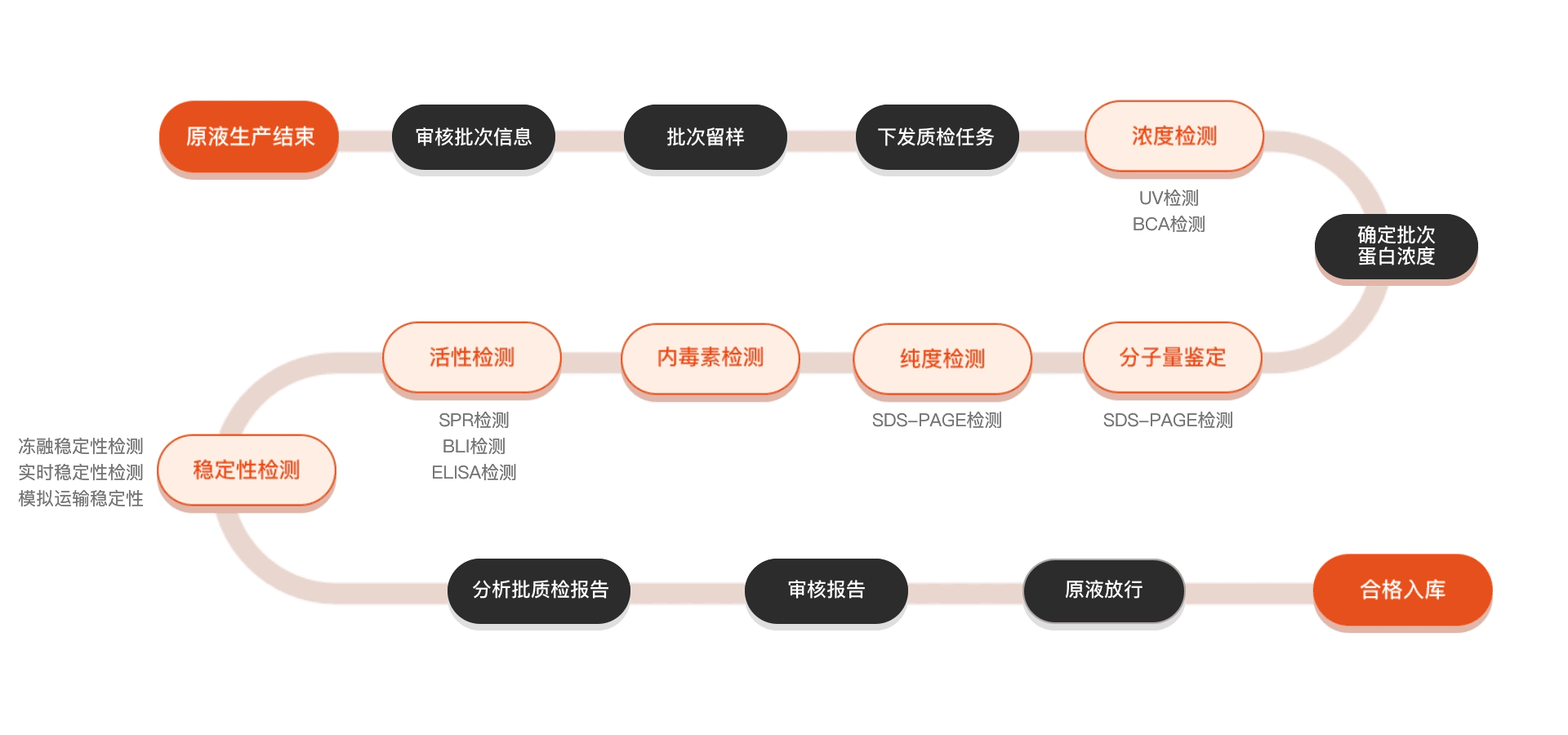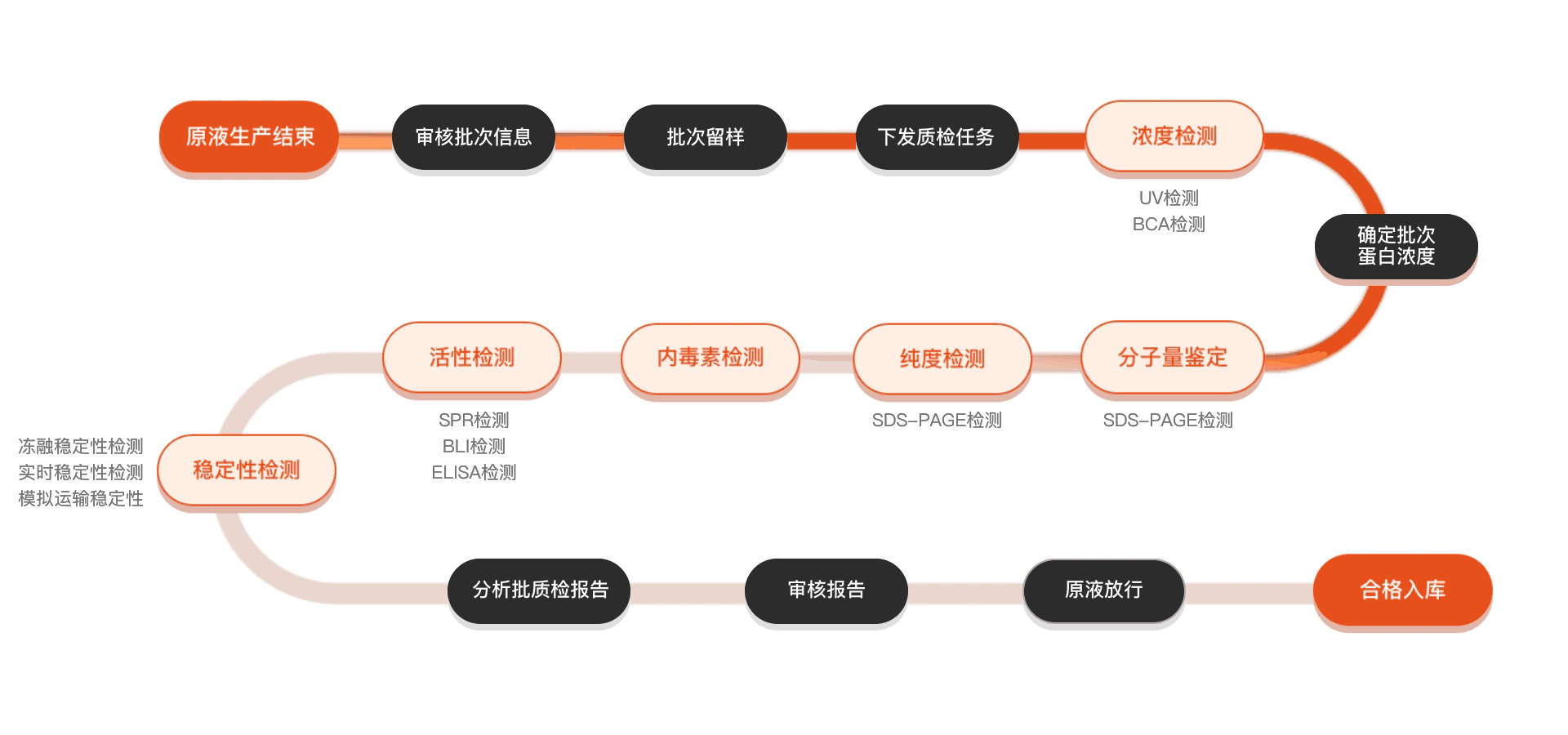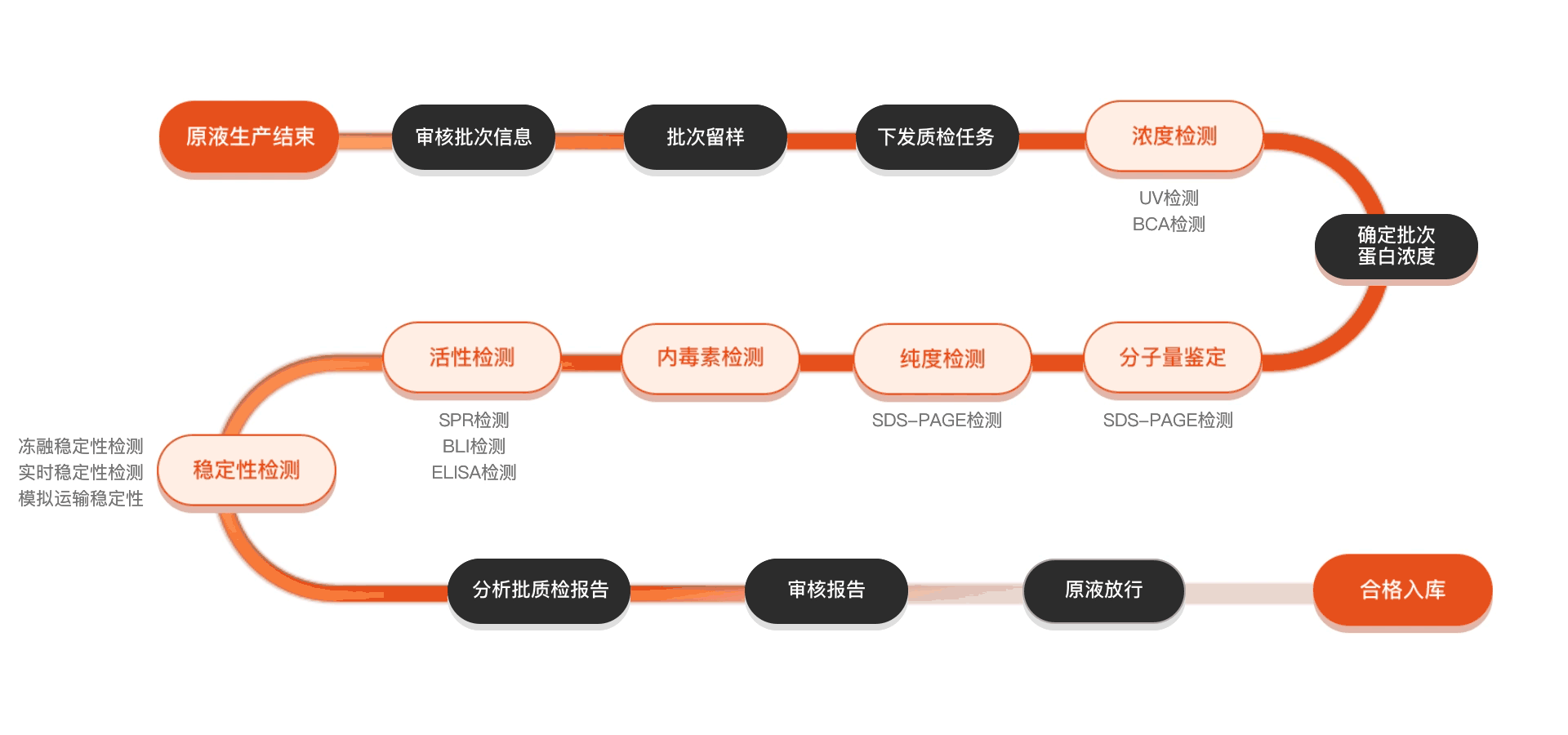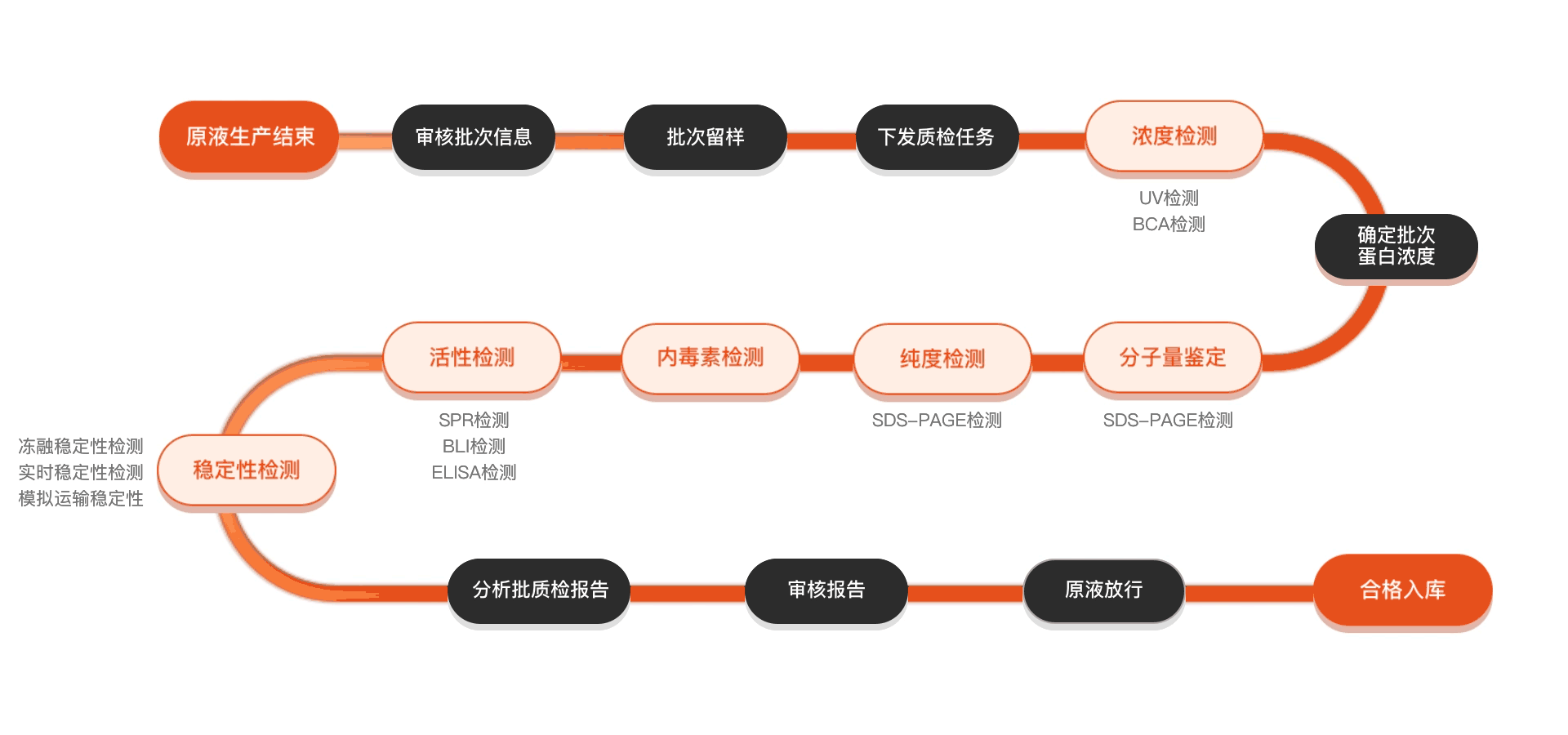
质检流程
相关产品






背景信息
RPL13, a key member of the ribosomal protein family, plays a crucial role in the assembly and function of the ribosome, which is essential for protein synthesis in all living organisms. Research on RPL13 has gained significant attention due to its involvement in various cellular processes and its implications in human diseases, especially cancer. Aberrations in ribosomal proteins, including RPL13, have been linked to ribosomopathies, a group of disorders characterized by defects in ribosome biogenesis and function. Additionally, recent studies have suggested that RPL13 may play a role in regulating cell proliferation and apoptosis, thereby influencing tumorigenesis. Given the foundational role of RPL13 in ribosome function and its potential involvement in disease mechanisms, the study of RPL13 recombinant proteins has become increasingly important. Researchers utilize recombinant techniques to produce RPL13 for structural and functional analyses, aiming to elucidate its role in ribosome assembly and its interaction with various biomolecules. Understanding the structure-function relationship of RPL13 could provide insights into its regulatory mechanisms and help identify potential therapeutic targets for diseases associated with ribosome dysfunction. As such, the investigation of RPL13 and its recombinant forms represents a critical area of research with the potential to advance our understanding of fundamental biological processes and their implications in health and disease.